Amyloidosis Treatment in india
Amyloidosis is a rare and serious condition caused by the abnormal accumulation of amyloid proteins in various organs, including the heart, kidneys, liver, and nerves. If left untreated, it can lead to progressive organ failure. The disease has multiple types—AL, ATTR, AA, and others—each requiring highly specialized care based on the underlying protein involved and organ damage present.
India has become a preferred destination for international patients seeking affordable, advanced care for amyloidosis. While diagnosis and initial workup typically cost between $800–$1,500, full chemotherapy for AL amyloidosis may cost $3,000–$7,000. In more advanced cases, autologous stem cell transplant (ASCT) is offered in India for $20,000–$30,000, compared to $300,000+ in the USA. For ATTR amyloidosis, modern drugs like Tafamidis and Patisiran are available at $1,000–$4,000/month, significantly more affordable than in Western countries.
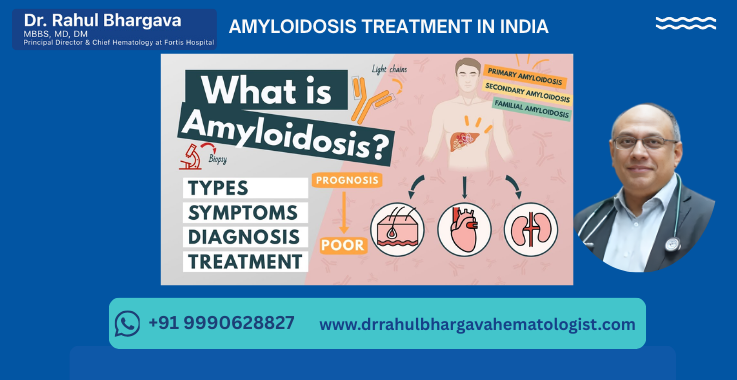
What is Amyloidosis?
Amyloidosis is a rare and serious disease that occurs when an abnormal protein, called amyloid, builds up in organs and tissues throughout the body. This accumulation can interfere with normal function and cause damage. The amyloid deposits can affect various organs, including the heart, kidneys, liver, spleen, nervous system, and digestive tract.
What are the Types of Amyloidosis and Specific Treatment?
1. AL (Light Chain) Amyloidosis:
AL amyloidosis is the most common type, resulting from the overproduction of light chains by abnormal plasma cells.
Chemotherapy and Targeted Therapy:
- Chemotherapy (for Plasma Cell Dyscrasias):
- Cyclophosphamide + Bortezomib + Dexamethasone (CyBorD regimen): This combination is commonly used for patients with AL amyloidosis, aiming to reduce the production of amyloid-forming light chains.
- Lenalidomide + Dexamethasone: An alternative to CyBorD, particularly for patients who are not candidates for stem cell transplant.
- Bortezomib (Velcade): A proteasome inhibitor that targets the plasma cells producing amyloid-forming proteins.
- Autologous Stem Cell Transplant: Considered for eligible patients with good performance status and organ function.
- Immunotherapy: Daratumumab, a monoclonal antibody targeting CD38, is used for patients who have relapsed or are refractory to other therapies.
2. AA (Secondary) Amyloidosis:
AA amyloidosis is caused by the deposition of amyloid fibrils derived from serum amyloid A (SAA), typically associated with chronic inflammatory conditions such as rheumatoid arthritis or chronic infections.
Treating the Underlying Disease:
- Corticosteroids: For patients with chronic inflammatory diseases, corticosteroids (e.g., prednisone) can help reduce SAA levels and amyloid deposition.
- Disease-Modifying Antirheumatic Drugs (DMARDs): Medications such as methotrexate or biologics (e.g., TNF inhibitors like infliximab) can be used to manage the underlying inflammatory condition.
- Interleukin-1 inhibitors, such as anakinra and canakinumab, are being explored to reduce the production of SAA in patients with chronic inflammation.
3. Familial (Hereditary) Amyloidosis:
Familial amyloidosis results from mutations in the transthyretin (TTR) gene, leading to the accumulation of amyloid deposits in organs like the heart and nerves.
- TTR Stabilisers:
- Tafamidis: An oral medication that stabilises the TTR protein and prevents amyloid formation. It is approved for patients with transthyretin amyloid cardiomyopathy.
- Diflunisal: Another TTR stabiliser that has shown some benefit in slowing progression.
- Gene Silencing Therapies:
- Patisiran and inotersen are RNA interference therapies that target the TTR gene, reducing the production of amyloid-forming proteins. These therapies are used for patients with hereditary ATTR amyloidosis.
- Liver Transplantation:
- For patients with severe familial amyloidosis, liver transplantation can be considered, as the liver is the source of the mutated TTR protein. A liver transplant can cure the condition but requires careful consideration of the patient’s overall health.
4. Dialysis-Related Amyloidosis:
This occurs in patients with long-term dialysis, often due to the accumulation of β2-microglobulin in tissues.
- Dialysis Modifications: Enhanced dialysis techniques can lower β2-microglobulin levels and slow amyloid deposition.
- Bisphosphonates: Medications like pamidronate can help manage bone-related complications in dialysis-related amyloidosis.
| Type | Protein Involved | Cause / Associated Condition | Common Organs Affected |
|---|---|---|---|
| 1. AL (Light-Chain) Amyloidosis | Immunoglobulin light chains | Plasma cell disorders (e.g., multiple myeloma) | Heart, kidneys, liver, nerves |
| 2. AA (Secondary) Amyloidosis | Serum amyloid A | Chronic inflammatory diseases (e.g., RA, TB, IBD) | Kidneys, liver, spleen |
| 3. ATTR (Transthyretin) Amyloidosis | Transthyretin (TTR) | Hereditary mutations or age-related changes in TTR protein | Heart, peripheral nerves |
| – Hereditary ATTR (hATTR) | Mutant TTR | Genetic (autosomal dominant inheritance) | Heart, nerves, eyes |
| – Wild-Type ATTR (wtATTR) | Normal TTR | Age-related degeneration (mainly in elderly men) | Heart (restrictive cardiomyopathy) |
| 4. Dialysis-Related Amyloidosis | β2-microglobulin | Long-term hemodialysis (>5 years) | Joints, bones, tendons |
| 5. ALECT2 Amyloidosis | Leukocyte chemotactic factor 2 | Rare; genetic predisposition, especially in some ethnicities | Kidneys, liver |
| 6. Apolipoprotein-Related | ApoAI, ApoAII, ApoAIV | Rare hereditary types | Kidneys, liver, heart |
| 7. Gelsolin Amyloidosis | Gelsolin | Rare hereditary disease | Eyes, skin, cranial nerves |
Quick Summary
| Type | Hereditary? | Treatable/Curable? | Main Treatment Options |
|---|---|---|---|
| AL | ❌ | Potentially curable | Chemotherapy, stem cell transplant |
| AA | ❌ | Treatable | Control underlying inflammation |
| Hereditary ATTR | ✅ | Treatable, not curable | Tafamidis, Patisiran, Inotersen, liver transplant (some) |
| Wild-type ATTR | ❌ | Treatable | Tafamidis, supportive cardiac care |
| Dialysis-related | ❌ | Treatable | Kidney transplant, dialysis modification |
| ALECT2 | ❌ | Limited treatment | Organ support (no specific targeted therapy yet) |
| Apolipoprotein types | ✅ | Supportive care | Rare; case-by-case management |
Diagnosis & Evaluation
- Blood tests: CBC, kidney & liver function, calcium, BNP, troponin
- Urine tests: 24-hour protein, Bence Jones protein
- Serum Free Light Chain (sFLC) assay
- Electrophoresis (SPEP, UPEP) with immunofixation
- Tissue Biopsy: Fat pad, bone marrow, or affected organ
- Bone Marrow Biopsy: Especially for AL amyloidosis
- Echocardiogram, Cardiac MRI, SAP scan: For cardiac involvement
- Genetic testing (for hereditary ATTR)
Treatment Protocol by Type
A. AL Amyloidosis (Light Chain)
Goal: Suppress abnormal plasma cells producing light chains
- First-line Therapy:
- Bortezomib + Cyclophosphamide + Dexamethasone (CyBorD)
- Add Daratumumab (anti-CD38 monoclonal antibody) if advanced
- Autologous Stem Cell Transplant (ASCT):
- For eligible patients (young, good organ function)
- High-dose Melphalan followed by stem cell infusion
- Supportive care: Diuretics, anticoagulants, dialysis if renal failure
B. ATTR Amyloidosis (Transthyretin-related)
Goal: Stabilise or reduce transthyretin amyloid deposits
- Medications:
- Tafamidis (TTR stabilizer – FDA approved for cardiac ATTR)
- Patisiran or Inotersen (gene silencers for hereditary ATTR)
- Liver Transplant: For hereditary ATTR
- Supportive Care: Pacemakers for cardiac block, pain relief for neuropathy
C. AA Amyloidosis (Secondary)
Goal: Treat underlying inflammatory disease
- Anti-inflammatory Treatment: Biologics like TNF inhibitors in RA, or antibiotics for TB
- Colchicine: For Familial Mediterranean Fever
- Organ support: Nephrology for proteinuria, renal function
D. Dialysis-Related Amyloidosis
- Optimise dialysis techniques (high-flux membranes)
- Kidney Transplant: May halt amyloid progression
- Pain management and orthopaedic surgery if bone/joint involvement
Amyloidosis Treatment Cost Comparison: India vs Turkey vs USA
| Treatment Type | India (USD) | Turkey (USD) | USA (USD) |
|---|---|---|---|
| Diagnostic Workup (Blood, Biopsy, Imaging) | $800 – $1,500 | $2,000 – $3,500 | $5,000 – $10,000 |
| AL Amyloidosis Chemo (CyBorD ± Daratumumab) | $3,000 – $7,000 (6 cycles) | $8,000 – $15,000 | $30,000 – $60,000 |
| Autologous Stem Cell Transplant (ASCT) | $20,000 – $30,000 | $40,000 – $60,000 | $300,000 – $400,000 |
| Tafamidis (ATTR, per month) | $1,000 – $1,800/month | $2,500 – $4,000/month | $20,000 – $25,000/month |
| Patisiran/Inotersen (per month) | $2,500 – $4,000/month | $5,000 – $8,000/month | $35,000 – $45,000/month |
| Liver Transplant (for hereditary ATTR) | $18,000 – $30,000 | $40,000 – $70,000 | $300,000+ |
| Kidney Transplant (AA or dialysis-related) | $12,000 – $18,000 | $30,000 – $45,000 | $250,000 – $350,000 |
| Dialysis (annual cost) | $2,000 – $4,000 | $8,000 – $12,000 | $50,000 – $80,000 |
| Supportive Cardiac/Renal Care (annual) | $1,000 – $2,500 | $3,000 – $5,000 | $15,000 – $25,000 |
| Post-Treatment Monitoring (6–12 months) | $500 – $1,200 | $1,500 – $3,000 | $10,000 – $20,000 |
Key Insights:
- India offers world-class amyloidosis care (including ASCT and ATTR-targeted drugs) at 70–90% lower costs than the USA.
- Turkey provides mid-tier affordability with modern infrastructure.
- USA costs remain prohibitively high, especially for rare disease treatments and transplants.
Amyloidosis Recovery Period
The recovery timeline in amyloidosis depends on the type of amyloidosis, organs affected, and the treatment received — such as chemotherapy, stem cell transplant, or targeted therapies. Since organ damage may already be present at diagnosis, recovery involves both stopping disease progression and supporting organ function.
1. Recovery After Chemotherapy (AL Amyloidosis)
| Phase | Timeline | Details |
|---|---|---|
| Induction Period | 4–6 months | Blood light chain levels monitored. Side effects subside gradually. |
| Hematologic Response | 3–6 months | Free light chain levels normalize; organ function may begin improving. |
| Organ Response | 6–12 months | Kidney, heart, or liver function gradually improves. |
| Maintenance/Remission | After 12 months | Regular monitoring; long-term remission possible. |
📝 Note: Organ improvement often lags behind blood response by several months.
2. Recovery After Autologous Stem Cell Transplant (ASCT)
| Phase | Timeline | What Happens |
|---|---|---|
| Hospitalization | 3–4 weeks | High-dose chemo + stem cell infusion. Neutropenia and fatigue. |
| Blood Cell Recovery | 2–4 weeks | WBC, platelets, hemoglobin begin to rise. |
| Functional Recovery | 3–6 months | Energy, appetite improve. Organ function monitored. |
| Immune System Stabilization | 6–12 months | Immunizations resume; fatigue resolves. |
| Long-Term Remission | 1+ year | Sustained remission in well-selected patients. |
✅ Best outcomes in patients with early-stage AL amyloidosis and minimal organ damage.
3. Recovery After Tafamidis, Patisiran, or Inotersen (ATTR Amyloidosis)
| Phase | Timeline | What to Expect |
|---|---|---|
| Symptom Stabilization | 1–3 months | Slowing of disease progression; fewer hospitalizations. |
| Functional Improvement | 3–6 months | Improved walking, less neuropathy or shortness of breath. |
| Ongoing Therapy | Lifelong | Continuous therapy needed to sustain benefit. |
🧠 Recovery is gradual, and the goal is stabilization or modest improvement, not cure.
4. Recovery After Liver/Kidney Transplant (ATTR or Dialysis-Related Types)
| Phase | Timeline | Details |
|---|---|---|
| Hospitalization | 2–3 weeks | Post-surgery ICU + ward stay |
| Initial Recovery | 1–3 months | Immunosuppression; infection risk monitoring |
| Functional Gains | 3–6 months | Organ function normalizes (creatinine, liver enzymes) |
| Full Recovery | 6–12 months | Return to normal life, provided no complications |
General Recovery Tips
- Diet: Low-sodium, high-protein (especially for kidney or cardiac involvement)
- Compliance: Strict adherence to medications (chemo, immunosuppressants, targeted therapies)
- Activity: Gradual return to physical activity, based on cardiac/kidney function
- Follow-up: Regular monitoring of free light chains, NT-proBNP, creatinine, and drug levels
Frequently Asked Questions
Commonly affected organs include the kidneys, heart, liver, spleen, nerves, and gastrointestinal tract.
AL amyloidosis is caused by the abnormal production of light chain proteins by plasma cells, which form amyloid deposits. It is often associated with multiple myeloma.
AA amyloidosis is secondary to chronic inflammatory diseases, infections, or malignancies and involves the deposition of serum amyloid A protein.
Hereditary amyloidosis is caused by inherited mutations in specific genes, leading to abnormal amyloid protein production. Transthyretin (TTR) amyloidosis is the most common form.
Wild-type ATTR amyloidosis occurs due to the accumulation of normal transthyretin protein in elderly individuals, primarily affecting the heart.
Dialysis-related amyloidosis is caused by the accumulation of beta-2 microglobulin in patients undergoing long-term dialysis.