Sickle Cell Anemia Treatment in India
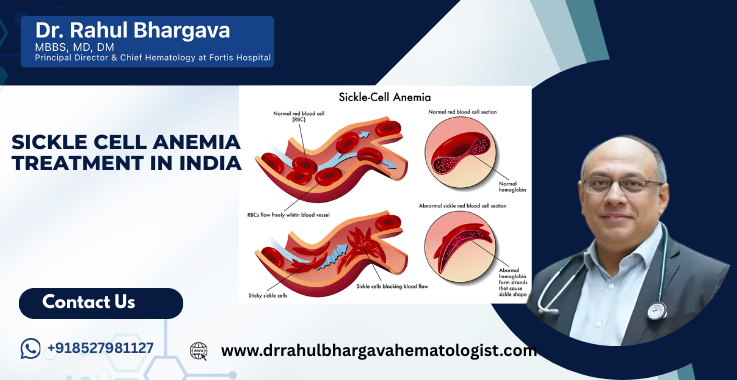
Sickle cell anemia is a severe genetic blood disorder that affects millions of people worldwide, especially in regions such as Africa, the Middle East, South Asia, and parts of India. The disease changes the shape and function of red blood cells, leading to anemia, chronic pain, and serious health complications. For many families, navigating treatment options and costs can be overwhelming, especially when considering care outside their home country.
India is a leading destination for affordable and advanced treatment for Sickle Cell Anemia. Importantly, the cost of sickle cell anemia treatment in India ranges from ₹5,00,000 to ₹25,00,000 ($6,000–$30,000) depending on the treatment plan, especially if a stem cell transplant is involved. With world-class hospitals, experienced hematologists, and access to life-saving procedures like bone marrow transplants, international patients are increasingly choosing India for both quality and value.
What is Sickle Cell Anemia?
Sickle cell anemia is a chronic, inherited blood disorder caused by a gene mutation. The gene is responsible for instructing the body on how to produce hemoglobin (the protein in red blood cells that transports oxygen). In people with this condition, the red blood cells become sticky, hard, and shaped like a sickle. These sickle cells die prematurely, blocking blood flow and leading to pain, infections, organ damage, and fatigue.
It is one of several conditions under the umbrella of sickle cell disease (SCD). It is an autosomal recessive condition, meaning a child must inherit the mutated gene from both parents to develop the disease. If only one parent passes on the gene, the child becomes a carrier (known as Sickle Cell Trait) but usually does not show symptoms.
Key Characteristics of Sickle Cell Anemia:
- Red blood cells lose their standard disc shape and flexibility.
- Abnormal cells clump together and block blood flow in blood vessels.
- Blockages can cause pain, strokes, and damage to vital organs like the spleen, liver, and kidneys.
- Red cells break down faster than usual, causing persistent anemia.
Global and Regional Burden:
According to the World Health Organization (WHO), over 300,000 children are born annually with SCD, with the highest prevalence in sub-Saharan Africa, India, and the Middle East. In India, it is particularly common among tribal populations in states such as Maharashtra, Odisha, Chhattisgarh, Madhya Pradesh, and Gujarat.
Impact on Daily Life:
- Children and adults may experience episodes of extreme pain called sickle cell crises.
- Fatigue and shortness of breath are common symptoms that are associated with low levels of red blood cells.
- There is a higher risk of infections, especially if the spleen is damaged or removed.
- School and work attendance may suffer due to frequent hospital visits or complications.
What are the Different Types of Sickle Cell Anemia?
Sickle cell anemia is part of a broader group known as sickle cell disease (SCD). Not all types are the same, and their severity varies depending on the type of gene mutation inherited. Understanding the different types helps doctors and families choose the right treatment strategy and set realistic expectations for outcomes. The main types are:
- HbSS (Sickle Cell Anemia): This is the most common and severe form of SCD. Patients inherit two sickle cell genes (one from each parent). It causes chronic anemia, pain crises, and severe organ complications. Most of the content in this article refers to this type unless otherwise stated.
- HbSC Disease: It occurs when a person inherits one sickle cell gene and one gene for hemoglobin C, a different abnormal hemoglobin. It generally causes milder symptoms than HbSS but can still lead to complications such as vision problems and joint pain.
- HbS Beta-Thalassemia (Sβ⁰ or Sβ⁺): This type occurs when one sickle cell gene is inherited along with a beta-thalassemia gene. The severity of symptoms depends on whether the beta-thalassemia gene is mild (Sβ⁺) or severe (Sβ⁰). People with Sβ⁰ thalassemia often have symptoms similar to HbSS.
- HbSD, HbSE, and HbSO: These are rarer forms of SCD and result from combinations of the sickle gene with other abnormal hemoglobins. The clinical features can vary widely, and diagnosis often requires detailed lab testing and genetic counseling.
What are the Symptoms of Sickle Cell Anemia?
The symptoms of sickle cell anemia can vary in intensity, frequency, and pattern depending on the individual's age, the type of sickle cell disease they have, and their overall health. Symptoms typically begin in early childhood and progress over time.
Common Symptoms Seen in Children and Adults
- Anemia-Related Fatigue: Chronic anemia leads to persistent tiredness, low stamina, paleness, and difficulty concentrating due to the reduced number of healthy red blood cells.
- Pain Episodes (Sickle Cell Crises): One of the most defining and painful features of the disease, these episodes occur when sickle-shaped cells block blood flow. Pain can be sharp or dull and usually affects the chest, abdomen, joints, or limbs. It can last from a few hours to several days.
- Swelling in Hands and Feet (Dactylitis): In infants and children, reduced circulation in the small bones can cause painful swelling, often misinterpreted as an infection or injury.
- Frequent Infections: Sickle cells damage the spleen, an organ that plays a crucial role in fighting infections. Children are especially vulnerable to pneumonia, meningitis, and other severe diseases. Preventive antibiotics and vaccinations are essential.
- Delayed Growth and Puberty: Poor oxygen delivery to tissues and chronic anemia can cause delays in height and weight gain in children. Puberty may also occur later than average.
- Vision Problems: Blood vessels in the eyes become blocked, leading to damage to the retina. If untreated, it can cause partial or complete vision loss.
- Shortness of Breath and Chest Pain: A life-threatening complication known as acute chest syndrome may occur when sickled cells block vessels in the lungs. Symptoms include cough, fever, low oxygen levels, and shortness of breath.
- Jaundice and Yellowing of the Eyes: The rapid breakdown of RBCs can lead to an overload of bilirubin, resulting in yellowing of the eyes and skin.
- Leg Ulcers and Skin Wounds: Chronic poor circulation can lead to non-healing wounds, especially around the ankles or lower legs.
- Stroke or Transient Ischemic Attacks (TIAs): Children and young adults with severe SCD may suffer from strokes due to blocked vessels in the brain. Regular transcranial Doppler screening can help detect high-risk.
Variation in Symptoms
- Infants (0–1 year): May show hand-foot swelling, irritability, and delayed growth.
- Children (1–12 years): Frequent pain crises, fatigue, learning difficulties, and increased infections.
- Adolescents & Adults: More pronounced episodes of pain, joint damage, fertility issues, and risk of organ failure.
Chronic Complications to Watch For
- Kidney damage (protein in urine)
- Gallstones
- Liver enlargement or damage
- Avascular necrosis (joint damage, especially in the hips)
- Heart problems due to chronic anemia
Emotional and Social Impact
Chronic pain, hospitalizations, and fatigue can lead to school absences, job instability, and emotional distress. Patients may struggle with anxiety, depression, or feelings of isolation due to long-term illness.
Being familiar with these symptoms enables early intervention and better disease control. Comprehensive management, preventive care, and patient education play a key role in improving long-term quality of life.
How is Sickle Cell Anemia Diagnosed?
Early and accurate diagnosis of sickle cell anemia is essential for effective disease management. Advances in medical testing now allow for screening even before birth, helping families and healthcare providers prepare for the child's care needs from infancy.
Newborn Screening
In many countries, including India, newborn screening programs have started identifying children with sickle cell disease shortly after birth. It involves taking a few drops of blood from the baby's heel (heel prick test) to test for hemoglobin variants.
Confirmatory Tests
- Hemoglobin Electrophoresis: This is the most common and reliable test. It separates and identifies different types of hemoglobin in the blood. It can differentiate between sickle cell anemia (HbSS), sickle cell trait (HbAS), and other variants of sickle cell disease.
- High-Performance Liquid Chromatography (HPLC): A highly sensitive method that quantifies different types of hemoglobin. It is often used in combination with electrophoresis for precise diagnosis.
- DNA Testing: Genetic testing can detect specific mutations in the hemoglobin gene. It is beneficial in prenatal diagnosis and in distinguishing rare types, such as HbSD or HbSE.
- Complete Blood Count (CBC): A general blood test that often shows low hemoglobin, increased reticulocyte count (immature red blood cells), and abnormal red cell morphology.
- Peripheral Blood Smear: Microscopic examination of blood showing characteristic sickle-shaped cells, target cells, and Howell-Jolly bodies.
- Solubility Test: A screening test used to detect the presence of hemoglobin S. If positive, more specific confirmatory tests are needed.
Prenatal Diagnosis
- Amniocentesis and Chorionic Villus Sampling (CVS) allow for the detection of the sickle cell gene mutation in a fetus.
- Genetic counseling is recommended for couples where both partners are carriers (HbAS).
Additional Tests to Assess Disease Severity
Doctors may also order:
- Liver and kidney function tests
- Chest X-rays or lung scans
- Transcranial Doppler ultrasound (in children to assess stroke risk)
- MRI or CT scans for neurological symptoms
Importance of Early Diagnosis
Early detection enables:
- Prophylactic treatments like penicillin and vaccinations
- Parent and patient education
- Planning for regular follow-ups and treatment milestones
- Consideration of curative therapies like bone marrow transplant before severe complications develop
How is Sickle Cell Anemia Treated?
Sickle cell anemia is a lifelong condition with no universal cure; however, several treatment strategies help manage symptoms, prevent complications, and improve survival and quality of life. The choice of treatment for sickle cell anemia depends on the patient's age, disease severity, organ involvement, and access to advanced care options.
Preventive Care and Health Maintenance
A significant part of managing Sickle Cell Anemia involves preventing complications before they happen. This is especially critical for infants and children, who are vulnerable to infections and other early complications.
- Newborn prophylaxis: Infants diagnosed through screening are started on daily penicillin to prevent infections, such as pneumonia, which can be life-threatening.
- Vaccinations: Patients receive routine immunizations and additional vaccines (pneumococcal, meningococcal, and Haemophilus influenzae) to reduce the risk of infection.
- Nutrition and supplements: Folic acid is prescribed to help produce new red blood cells. A good, balanced diet and proper hydration are essential to avoid crises.
Medications
- Hydroxyurea: This is a disease-modifying drug that helps reduce the frequency of painful crises, acute chest syndrome, and the need for blood transfusions. It works by increasing fetal hemoglobin (HbF), which prevents red cells from sickling.
- It is widely used in both children and adults.
- Side effects may include nausea, lowered white blood cell count, or temporary hair loss.
- L-glutamine (Endari): Approved in certain countries, this oral amino acid supplement helps reduce oxidative stress in red blood cells and lowers the frequency of pain episodes.
- Voxelotor: A newer drug that improves hemoglobin's ability to carry oxygen and reduces hemolysis. It helps manage anemia.
- Crizanlizumab: A monoclonal antibody that targets P-selectin, reducing inflammation and sickle cell adherence to vessel walls. Administered intravenously once a month.
Pain Management
Pain episodes (sickle cell crises) are treated with hydration, oxygen therapy, NSAIDs, and sometimes opioids for severe pain. Chronic pain management may require physical therapy, counseling, and long-term medications.
Blood Transfusions
Regular or emergency transfusions are often required to treat:
- Severe anemia
- Acute chest syndrome
- Stroke prevention or treatment
- Preparation for surgeries
While effective, frequent transfusions may lead to iron overload, requiring iron-chelation therapy.
Bone Marrow (Stem Cell) Transplantation
Currently, this is the only curative treatment for Sickle Cell Anemia.
- Autologous (self-derived) transplants are not effective in this condition.
- Allogeneic transplants from an HLA-matched sibling donor have shown high cure rates (up to 90%).
- Patients must be carefully selected, and transplants are typically recommended for those with severe complications or early signs of organ damage.
- India offers advanced transplant procedures at a significantly lower cost than Western countries.
Eligibility criteria include:
- Age usually under 16–18 years (though adult transplants are growing)
- Frequent pain crises or organ damage
- Availability of a matched sibling or matched unrelated donor
Gene Therapy and Research
Gene editing and stem cell manipulation are evolving areas of research. Although not yet widely available or affordable, early trials using technologies like CRISPR-Cas9 have shown promising results. India may see these options become viable in the next few years as technology and regulations evolve.
Supportive Therapies and Counseling
Chronic illness can affect mental health, education, and family life. Many Indian hospitals now offer:
- Counseling for depression, anxiety, and coping skills
- Educational support programs for children with frequent hospitalizations
- Vocational and genetic counseling for families and at-risk couples
What is the Cost of Sickle Cell Anemia Treatment in India?
India is globally recognized for offering world-class cancer treatment. The cost of sickle cell anemia treatment in India ranges from ₹5,00,000 to ₹25,00,000 ($6,000–$30,000). The price varies depending on the type of treatment, disease severity, hospital infrastructure, and the presence of complications.
Detailed Cost Breakdown
- The cost of routine medical management, including hydroxyurea therapy, pain medications, and regular doctor consultations, typically ranges between ₹15,000 and ₹30,000 per month, which is approximately $180 to $360 per month. On an annual basis, this amounts to ₹1.8 lakh to ₹3.6 lakh (or $2,100 to $4,300).
- Blood transfusion sessions, which are sometimes required monthly or during crises, cost around ₹10,000 to ₹25,000 per session. The cost ranges from $120 to $300 per session, depending on the hospital, the type of blood products used, and the level of supportive care required.
- Iron chelation therapy, which removes excess iron from the body after repeated transfusions, may cost between ₹40,000 and ₹1,20,000 per year, or approximately $500 to $1,500, depending on the medication and dosage.
- A bone marrow transplant (allogeneic) is currently the only curative option for sickle cell anemia and costs between ₹15,00,000 and ₹25,00,000. The price is equivalent to approximately $20,000 to $35,000 and includes the donor search or match, pre-transplant evaluations, hospital stay, transplant procedure, and post-operative care.
- Advanced or newer therapies, such as Crizanlizumab or Voxelotor (if available), can cost between ₹1,00,000 and ₹3,00,000 per month, which translates to $1,200 to $3,600 per month. These are not routinely used in all Indian hospitals due to their high cost and limited availability; however, they may be offered in top-tier private institutions.
Additional Costs to Consider
- The cost of accommodation for international patients and their families in guest houses or service apartments typically ranges between ₹1,500 and ₹4,000 per day, depending on the location and level of comfort desired.
- The pre-transplant evaluation, which includes HLA typing, organ function tests, and infection screening, costs around ₹75,000 to ₹1,50,000 or $900 to $1,800.
- After a successful transplant, post-transplant medications and follow-up care may require an annual budget of ₹1,00,000 to ₹2,00,000, which is approximately $1,200 to $2,400.
Patients from Europe, Africa, Asia, the Middle East, and neighboring countries often save tens of thousands of dollars while receiving care that matches international standards.
What are the Factors Affecting the Cost of Sickle Cell Anemia Treatment in India?
The overall cost of treating sickle cell anemia in India varies significantly based on several clinical, logistical, and institutional factors. Understanding these elements helps patients and their families plan financially and make informed choices.
- Type and Severity of Disease: The cost of treatment depends on whether the patient has sickle cell trait (a milder form) or full-blown sickle cell disease (HbSS or compound variants, such as HbSC). Patients with frequent pain crises, organ involvement, or stroke risk often need more intensive and costly care, such as monthly transfusions or bone marrow transplants.
- Type of Treatment Chosen: Basic management with hydroxyurea and pain relief costs significantly less than advanced therapies like stem cell transplantation or biologic drugs. Newer drugs such as Crizanlizumab or Voxelotor, though effective, come at a premium and are not subsidized in many settings.
- Hospital and Location: Treatment costs can vary depending on the city and whether the patient chooses a government hospital, a private multispecialty center, or an international hospital. Metro cities like Delhi, Mumbai, Chennai, and Bangalore offer advanced care but may be slightly more expensive than hospitals in tier-2 cities.
- Availability of Donor for Transplant: If a bone marrow transplant is considered, the availability of a matched sibling donor can reduce costs by eliminating the need for expensive donor searches and HLA typing. Matched unrelated donor (MUD) transplants are more costly due to the extended donor matching and graft preparation required for these procedures.
- Pre-existing Complications: Patients with kidney damage, stroke history, or lung involvement may require extended hospital stays and ICU care, increasing overall expenses. Special care, including dialysis or ventilator support, if necessary, increases the cost.
- Duration of Hospital Stay: Prolonged hospitalization due to complications, infection risk, or post-transplant rejection leads to higher accommodation, monitoring, and treatment costs.
- Supportive Medications and Aftercare: The long-term use of iron chelators, immunosuppressants (following BMT), or hormone therapy can increase the yearly treatment burden. Follow-ups, lab tests, and imaging studies also add to ongoing expenses.
Cost Comparison of Sickle Cell Anemia Treatment Across Countries
India stands out as one of the most affordable and high-quality destinations for treating sickle cell anemia. Here is a comparison of treatment costs for some of the most common procedures across different countries:
|
Treatment Type |
India |
USA |
UK |
UAE |
|
Hydroxyurea (1 Year) |
$2,100 – $4,300 |
$10,000 – $20,000 |
$8,000 – $15,000 |
$6,000 – $12,000 |
|
Blood Transfusion (per unit) |
$120 – $300 |
$1,000 – $2,500 |
$800 – $1,500 |
$500 – $1,000 |
|
Bone Marrow Transplant (BMT) |
$20,000 – $35,000 |
$150,000 – $300,000 |
$120,000 – $250,000 |
$100,000 – $180,000 |
|
Iron Chelation (1 Year) |
$500 – $1,500 |
$5,000 – $10,000 |
$4,000 – $9,000 |
$3,500 – $8,000 |
This comparison clearly shows that India offers costs 70% to 90% lower than those in developed countries while maintaining international standards of care.
Do Indian Hospitals Compromise on the Quality of Treatment?
No, Indian hospitals do not compromise on quality. In fact, India is recognized globally for its excellence in medical care, particularly in fields such as hematology, oncology, and stem cell transplantation. Here's why:
- Accredited Hospitals: Many Indian hospitals, such as the Fortis Memorial Research Institute in Gurgaon, are NABH and JCI-accredited, indicating adherence to international standards and protocols.
- Experienced Doctors: Indian hematologists and transplant specialists, such as Dr. Rahul Bhargava, have decades of experience treating complex blood disorders and cancers.
- Advanced Equipment: Hospitals are equipped with cutting-edge technology, including molecular diagnostics, 3D imaging, and BMT units.
- Comprehensive Care: Treatment is holistic, covering not just the disease but also nutrition, emotional health, and post-discharge care.
- Patient-Centric Approach: Customized treatment plans, multilingual communication, and family-inclusive counseling ensure a smooth recovery journey.
What are the Services Available for International Patients Seeking Sickle Cell Anemia Treatment in India?
India is a global hub for various treatments thanks to its combination of affordability, skilled professionals, and patient-friendly services. International patients traveling for Sickle Cell Anemia treatment can expect:
- Visa and Travel Assistance: Hospitals offer dedicated visa support letters, assistance with FRRO registration, and help with airport pickups.
- Language Interpreters: Translators fluent in Arabic, French, Swahili, and other languages ensure smooth communication.
- Accommodation Support: Options range from budget guest houses to serviced apartments near hospitals.
- Custom Meal Planning: The hospital's nutritionists work closely with international patients to ensure that meals are culturally appropriate.
- Personal Care Managers: A dedicated international patient coordinator guides you throughout your hospital journey, from pre-admission to discharge.
- Teleconsultation Services: Before traveling, patients can consult doctors online to assess suitability and cost estimates.
What is the Future of Sickle Cell Anemia Treatment in India?
The outlook for sickle cell anemia in India is brighter than ever. With government support, advances in research, and increased public awareness, India is well-positioned to lead in comprehensive care for this genetic disorder.
- Expansion of Bone Marrow Registries: More families now have access to matched unrelated donors, thanks to the growth of stem cell banks.
- Development of Affordable Gene Therapies: Trials are underway in top institutions to make curative gene editing options financially accessible.
- National Sickle Cell Mission: India's government has pledged to eliminate Sickle Cell Disease by 2047 through widespread screening and preventive care.
- Insurance Inclusion: Many insurance companies in India are now covering the management of chronic diseases, transplants, and medications.
- NGO Support: Organizations are stepping in to fund surgeries, drugs, and transportation for underprivileged families.
Patient Testimonials
Patient from Nigeria:
"I brought my daughter to India for a bone marrow transplant after struggling for years with crises back home. Fortis Hospital gave us a second chance at life. The team was kind, affordable, and brilliant."
— Amina O., Lagos
Patient from South Africa:
"We couldn't afford treatment locally. India provided us with access to the best care without overwhelming us with debt. The transplant was a success, and my son is now pain-free."
— Thabo N., Johannesburg
Patient from Iran:
"The moment we reached Delhi, everything from visa support to hospital coordination went smoothly. Dr. Rahul Bhargava was very welcoming, and we felt at home during a difficult time."
— Mina R., Tehran
Frequently Asked Questions
Yes, a bone marrow transplant offers a potential cure, especially in young patients with matched donors. Otherwise, it is managed with medications and supportive care.
A hospital stay is typically 3–4 weeks, with follow-up monitoring for 6–12 months, depending on the complications.
With modern treatments, many people with Sickle Cell Anemia can live into their 50s or longer. Early diagnosis and treatment are crucial in managing the disease and improving quality of life.
With modern treatment, many patients live into their 40s, 50s, or beyond, particularly when complications are well-managed.
Many hospitals accept insurance from international providers or assist with reimbursement documentation for foreign patients.
Transplantation is considered for patients with severe symptoms, stroke history, or organ damage, especially if a matched sibling donor is available.
Obtain medical clearance from the doctor in your home country, bring any relevant medical reports, stay well-hydrated during flights, and arrive a few days early to acclimate to the environment before treatment begins.
Sickle cell anemia occurs due to a genetic mutation in the hemoglobin gene. The mutation causes the production of abnormal hemoglobin, known as hemoglobin S.
Yes, sickle cell anemia is inherited when a child receives one abnormal hemoglobin gene from each parent.
Yes, sickle cell anemia can damage vital organs such as the lungs, kidneys, brain, liver, and heart due to blocked blood flow.
Doctors confirm the diagnosis of sickle cell anemia through a blood test called hemoglobin electrophoresis, which detects abnormal hemoglobin types.
No, people with sickle cell trait carry one abnormal gene but usually do not show symptoms, while those with the disease have two abnormal genes and experience symptoms.
Yes, many people with sickle cell anemia can have children. However, genetic counseling is recommended to understand the risks to offspring.
There are no specific foods to avoid, but patients should maintain a balanced diet, avoid dehydration, and limit alcohol to prevent crises.
Travel is generally safe, but patients should avoid extreme temperatures, stay hydrated, and consult their doctor before flying or visiting high-altitude areas.
Yes, sickle cell anemia increases the risk of stroke, especially in children. Regular screening and preventive transfusions can reduce this risk.
Yes, it is most common among people of African, Middle Eastern, Indian, and Mediterranean descent.