Beta Thalassemia Major Treatment in India
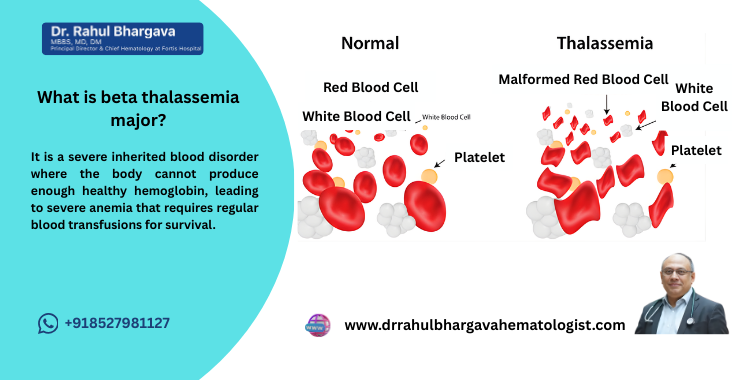
Beta Thalassemia Major, also known as Cooley’s Anemia, is a severe inherited blood disorder characterized by the inability to produce enough hemoglobin, the protein in red blood cells that carries oxygen throughout the body. This condition is caused by mutations in the HBB gene responsible for encoding the beta-globin chain of hemoglobin. Without adequate production of beta-globin chains, the structure of hemoglobin is disrupted, leading to ineffective red blood cell production and severe anemia.
Beta thalassemia major is the most severe form of thalassemia, and individuals with this condition often require frequent blood transfusions and ongoing medical management. It is typically diagnosed in early childhood.
What is Beta Thalassemia Major?
Beta Thalassemia Major, often referred to as Cooley’s Anemia, is the most severe form of thalassemia. Thalassemia is a group of inherited blood disorders that affect the body’s ability to produce hemoglobin, resulting in anemia. In Beta Thalassemia Major, both copies of the Beta-globin gene (one from each parent) are defective, leading to a lack of functional hemoglobin. This condition causes severe anemia, meaning the body
Types of Beta Thalassemia Major
Thalassemia is categorized into different types based on the severity of the condition. The two main types are:
1. Beta Thalassemia Major
This is the most severe form of thalassemia, where both Beta-globin genes are affected. Children born with this condition typically show symptoms in the first two years of life, which can range from pale skin, weakness, and fatigue to more severe complications like an enlarged spleen and liver, and growth delays.
2. Beta Thalassemia Intermedia
This is a milder form of Beta Thalassemia, where one of the Beta-globin genes is normal or only partially affected. People with Beta Thalassemia Intermedia may not require regular blood transfusions and may have fewer symptoms compared to Beta Thalassemia Major. However, the condition can still cause significant health issues, particularly if left untreated.
Causes of Beta Thalassemia Major
Beta Thalassemia Major is caused by genetic mutations in the HBB gene, which is responsible for producing Beta-globin, a component of hemoglobin. This gene mutation leads to insufficient production of hemoglobin, resulting in the characteristic symptoms of the disorder. The condition is inherited in an autosomal recessive manner, meaning a person must inherit a defective gene from both parents to develop the disease.
Symptoms of Beta Thalassemia Major
Beta Thalassemia Major usually presents in early childhood and requires immediate medical attention. Common symptoms include:
- Severe anemia: Fatigue, weakness, and pallor (pale skin).
- Growth Delays: Children with Beta Thalassemia Major may show delayed physical and developmental growth.
- Bone Deformities: The body compensates for anemia by producing more red blood cells, leading to bone changes (particularly in the face and skull).
- Enlarged Organs: An enlarged spleen (splenomegaly) and liver (hepatomegaly) due to increased blood cell destruction.
- Iron Overload: Due to frequent blood transfusions, excess iron accumulates in the body, leading to complications like heart disease and liver damage.
- Jaundice: Yellowing of the skin and eyes due to increased breakdown of red blood cells.
Diagnosis of Beta Thalassemia Major
Diagnosing Beta Thalassemia Major involves a combination of clinical evaluation, family history, and specific blood tests. Early diagnosis is essential for timely treatment and management.
Key Diagnostic Tests Include:
- Complete Blood Count (CBC): Measures red blood cell count, hemoglobin levels, and other important blood parameters.
- Hemoglobin Electrophoresis: This test identifies the types of hemoglobin in the blood and is crucial for detecting thalassemia.
- Genetic Testing: Confirmatory tests to identify mutations in the HBB gene and to determine whether one or both copies of the gene are affected.
- Iron Studies: To assess iron overload, which can occur due to frequent blood transfusions.
Treatment Options for Beta Thalassemia Major
While there is no complete cure for Beta Thalassemia Major in all cases, various treatment options are available to manage the symptoms and improve the quality of life.
1. Blood Transfusions
Regular blood transfusions are the cornerstone of treatment for Beta Thalassemia Major. These transfusions help to increase hemoglobin levels and relieve the symptoms of anemia. Depending on the severity of the condition, patients may need transfusions every 2-4 weeks.
2. Iron Chelation Therapy
Frequent blood transfusions can lead to iron overload in the body, which can damage organs. Iron chelation therapy helps to remove excess iron by using medications like deferasirox or deferoxamine. This treatment is crucial for preventing long-term complications.
Why Choose Dr. Rahul Bhargava for Beta Thalassemia Major Treatment?
Dr. Rahul Bhargava is one of India’s leading hematologists with vast expertise in the diagnosis and treatment of blood disorders like Beta Thalassemia Major. His clinic offers the latest advancements in medical care, ensuring patients receive the most effective and personalized treatments.
Reasons to Choose Dr. Rahul Bhargava:
- Experience: Dr. Bhargava has over [X] years of experience in hematology and has successfully treated numerous Beta Thalassemia patients.
- Advanced Care: His clinic utilizes state-of-the-art diagnostic tools and treatment options, including bone marrow transplants and iron chelation therapies.
- Patient-Centered Approach: Dr. Bhargava believes in a holistic, patient-first approach, ensuring emotional and physical support throughout the treatment journey.
Internationally Recognized: Dr. Bhargava is a recognized expert in hematology, with a global reputation for providing high-quality care for complex blood disorders.
Cost of Treatment for Beta Thalassemia Major in India
India offers affordable treatment for Beta Thalassemia Major, making it a popular destination for medical tourism. The costs of treatment vary based on the severity of the condition and the type of treatment required.
1. Blood Transfusions
- INR: ₹5,000 – ₹15,000 per session
- USD: $60 – $180 per session
2. Iron Chelation Therapy
- INR: ₹10,000 – ₹30,000 per month
- USD: $120 – $360 per month
3. Bone Marrow Transplant (BMT)
- INR: ₹10,00,000 – ₹25,00,000
- USD: $12,000 – $30,000
Frequently Asked Questions
Beta Thalassemia Major, also known as Cooley’s anemia, is a severe form of thalassemia where the body produces very little or no beta-globin, a critical component of hemoglobin. This leads to severe anemia and a lack of oxygen in the body.
It differs from Beta Thalassemia Minor, which is a carrier state with mild or no symptoms, and other forms of thalassemia (like alpha thalassemia), which affect different parts of the hemoglobin molecule.